Services
Real-Time Calculations
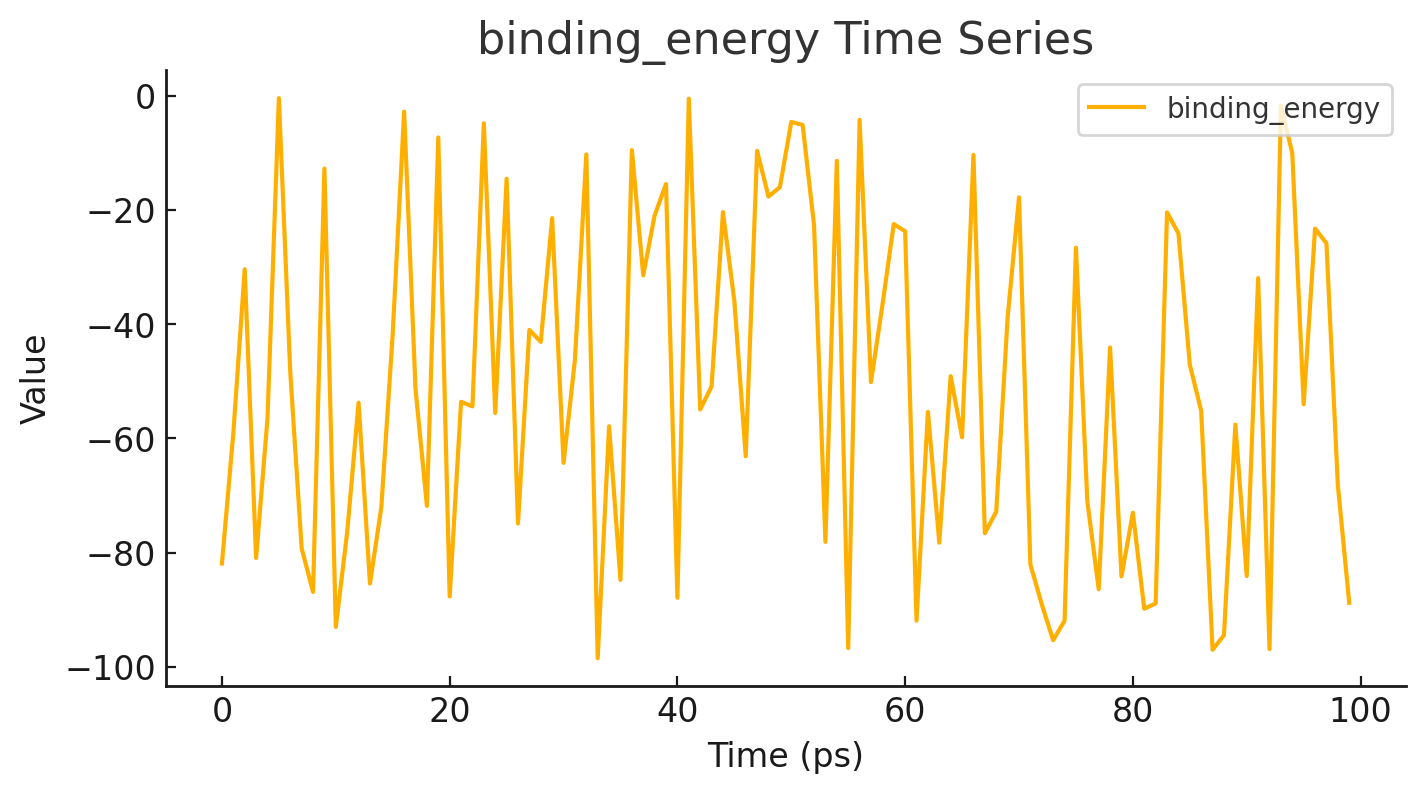
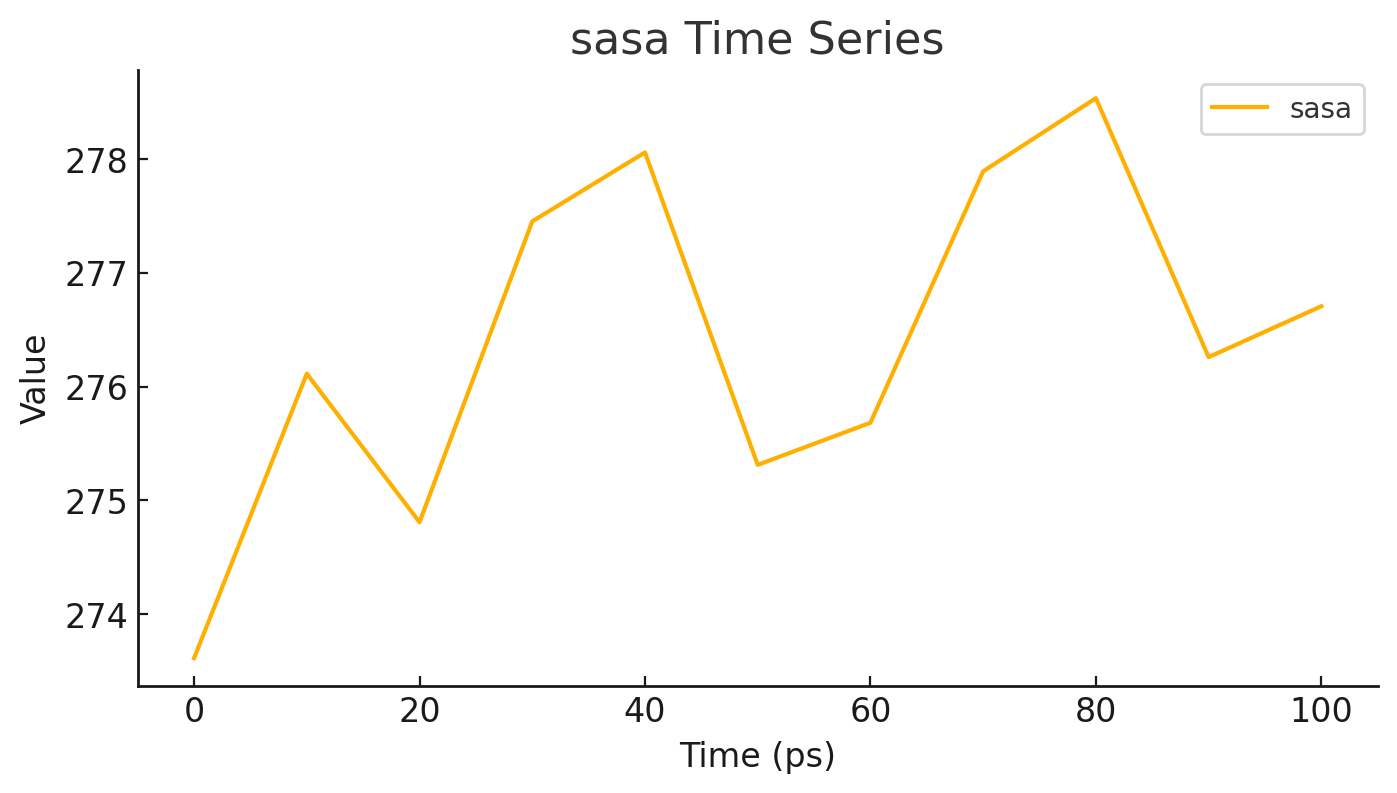
Calculate and visualize molecular interactions in real time.
Visualization
View molecular structures graphically.
Data Export
Export and analyze your simulation results in a variety of formats.
DOCKING Autodock Vine
docking predicts how a small molecule (ligand) binds to a target protein. The main steps are:
Prepare protein and ligand structures: The protein is kept rigid, while the ligand is flexible to explore binding poses.
Define the binding site: A 3D grid box is set around the target region where docking will occur.
Run docking calculations: Using algorithms like the Lamarckian Genetic Algorithm, the best ligand conformations are searched based on binding energy.
Analyze results: The lowest energy poses indicate the most likely binding modes.

Moleular Dynamics GROMACS
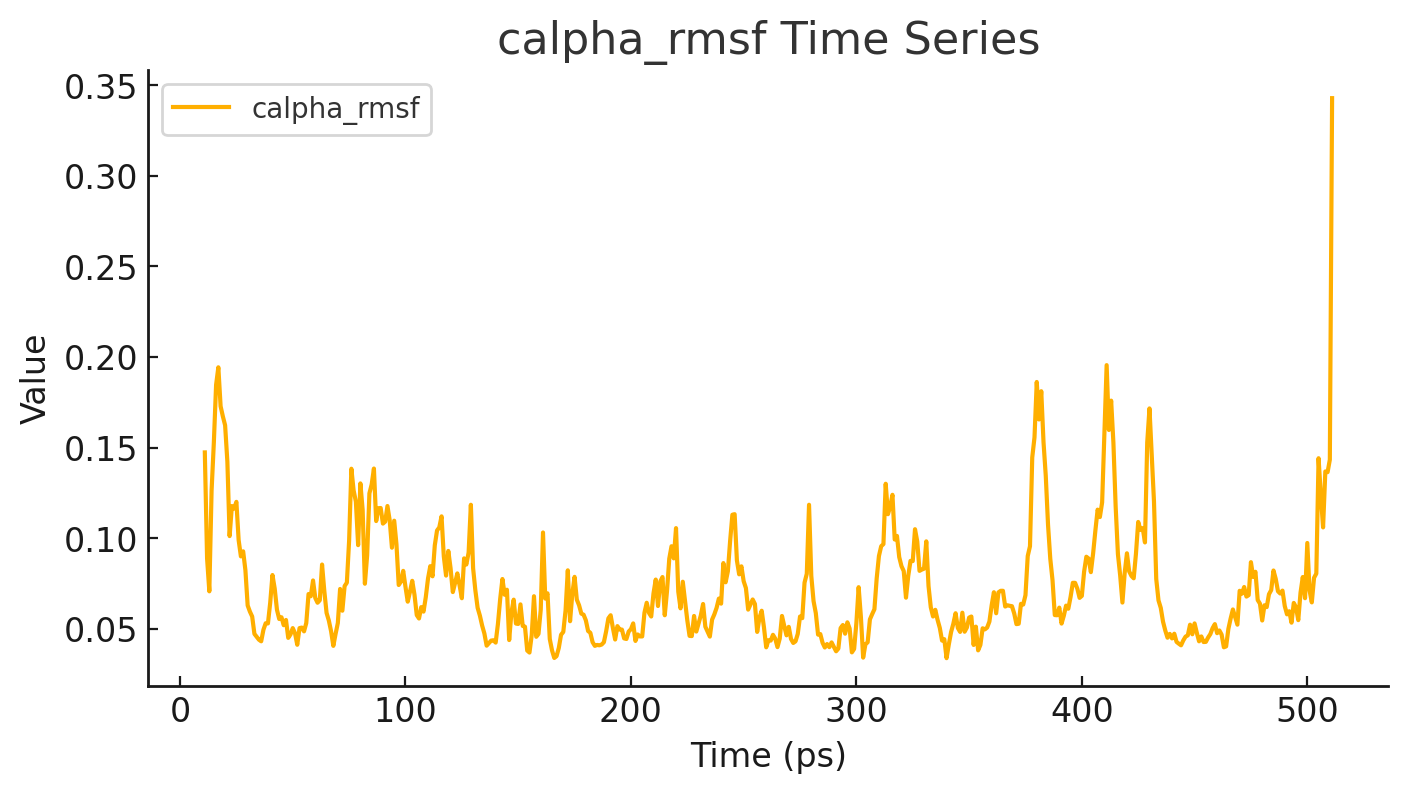
Molecular dynamics (MD) simulations with GROMACS involve preparing the biomolecular system, applying appropriate force fields, and running time-step integrations to model atomic motions over time; docking can be integrated by first predicting ligand binding poses using docking tools, then refining these complexes through MD to study their stability and dynamics in a realistic environment, making GROMACS a powerful platform for combined docking and simulation workflows.

Simulatin Files Filemail.
After the simulation is completed, all generated files are sent via Filemail for easy and secure sharing. The files remain available for download for one month, allowing you to access and retrieve your simulation data at your convenience within this period. This ensures that even large MD simulation files can be delivered quickly and efficiently.